Bagci, Vodovotz, Billiar, Ermentrout, Bahar, 2006
Model Status
This CellML model is able to run in PCEnv and COR to reproduce published results in the original paper. The units in this model have also been checked and curated.
Model Structure
ABSTRACT: We propose a mathematical model for mitochondria-dependent apoptosis, in which kinetic cooperativity in formation of the apoptosome is a key element ensuring bistability. We examine the role of Bax and Bcl-2 synthesis and degradation rates, as well as the number of mitochondrial permeability transition pores (MPTPs), on the cell response to apoptotic stimuli. Our analysis suggests that cooperative apoptosome formation is a mechanism for inducing bistability, much more robust than that induced by other mechanisms, such as inhibition of caspase-3 by the inhibitor of apoptosis (IAP). Simulations predict a pathological state in which cells will exhibit a monostable cell survival if Bax degradation rate is above a threshold value, or if Bax expression rate is below a threshold value. Otherwise, cell death or survival occur depending on initial caspase-3 levels. We show that high expression rates of Bcl-2 can counteract the effects of Bax. Our simulations also demonstrate a monostable (pathological) apoptotic response if the number of MPTPs exceeds a threshold value. This study supports our contention, based on mathematical modeling, that cooperativity in apoptosome formation is critically important for determining the healthy responses to apoptotic stimuli, and helps define the roles of Bax, Bcl-2, and MPTP vis-a-vis apoptosome formation.
The original paper is cited below:
Bistability in Apoptosis: Roles of Bax, Bcl-2 and Mitochondrial Permeability Transition Pores. E.Z. Bagci, Y. Vodovotz, T.R. Billiar et al, 2006, Biophysical Journal, 90, 1546-1559. PubMed ID: 16339882
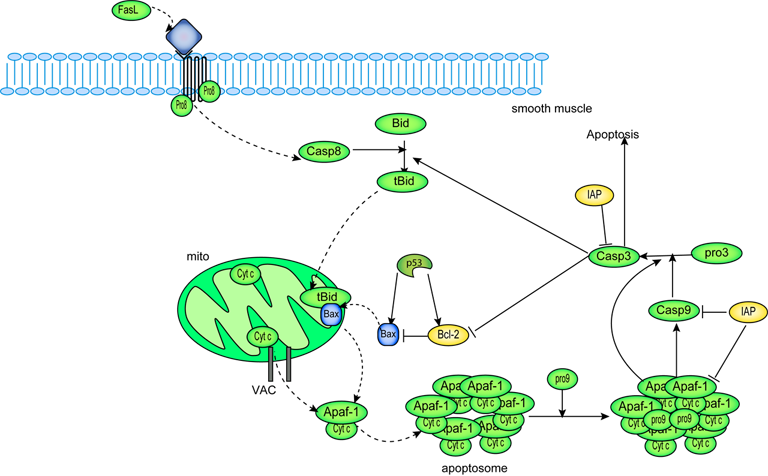 |
| A schematic diagram of intrinsic apoptotic signalling pathway described by this mathematical model. |