Priebe, Beuckelmann, 1998
Model Status
This CellML model runs in both COR and PCEnv to replicate the published results. The units have been checked and they are consistent.
Model Structure
ABSTRACT: Patients with severe heart failure are at high risk of sudden cardiac death. In the majority of these patients, sudden cardiac death is thought to be due to ventricular tachyarrhythmias. Alterations of the electric properties of single myocytes in heart failure may favor the occurrence of ventricular arrhythmias in these patients by inducing early or delayed afterdepolarizations. Mathematical models of the cellular action potential and its underlying ionic currents could help to elucidate possible arrhythmogenic mechanisms on a cellular level. In the present study, selected ionic currents based on human data are incorporated into a model of the ventricular action potential for the purpose of studying the cellular electrophysiological consequences of heart failure. Ionic currents that are not yet sufficiently characterized in human ventricular myocytes are adopted from the action potential model developed by Luo and Rudy (LR model). The main results obtained from this model are as follows: The action potential in ventricular myocytes from failing hearts is longer than in nonfailing control hearts. The major underlying mechanisms for this prolongation are the enhanced activity of the Na+-Ca2+ exchanger, the slowed diastolic decay of the [Ca2+]i transient, and the reduction of the inwardly rectifying K+ current and the Na+-K+ pump current in myocytes of failing hearts. Furthermore, the fast and slow components of the delayed rectifier K+ current (I(Kr) and I(Ks), respectively) are of utmost importance in determining repolarization of the human ventricular action potential. In contrast, the influence of the transient outward K+ current on APD is only small in both cell groups. Inhibition of I(Kr) promotes the development of early afterdepolarizations in failing, but not nonfailing, myocytes. Furthermore, spontaneous Ca2+ release from the sarcoplasmic reticulum triggers a premature action potential only in failing myocytes. This model of the ventricular action potential and its alterations in heart failure is intended to serve as a tool for investigating the effects of therapeutic interventions on the electric excitability of the human ventricular myocardium.
The original paper reference is cited below:
Simulation study of cellular electric properties in heart failure, Leo Priebe and Dirk J. Beuckelmann, 1998, Circulation Research, 82, 1206-1223. PubMed ID: 9633920
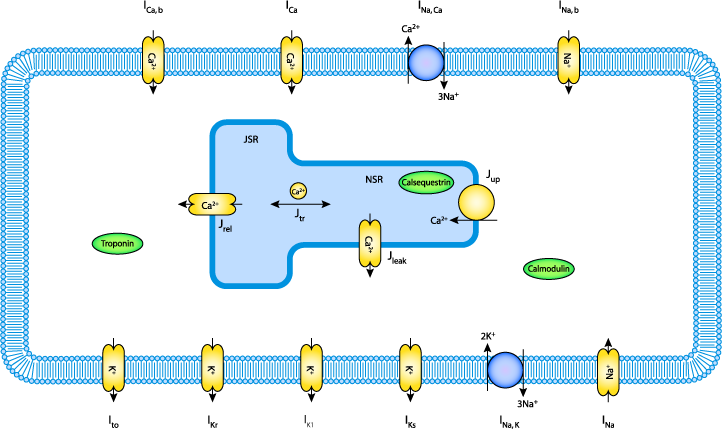 |
| A schematic diagram describing the transmembrane ionic currents through channels, pumps and exchangers, that are captured in the Priebe and Beuckelmann 1998 model of a human ventricular myocyte. The intracellular compartment in the diagram represents the sarcoplasmic reticulum (SR). This is divided into the two subcompartments, the network SR (NSR) and the junctional SR (JSR). Ca2+ buffers are present in both the cytoplasm and the JSR - troponin (TRPN), Calmodulin (CMDN), and calsequestrin (CSQN). |